CRISPR, the gene-editing technology that has revolutionized biological research, is finally available as a medical treatment with regulatory approval. On December 8 the U.S. Food and Drug Administration approved the first CRISPR treatment for sickle cell disease.
The treatment, called exa-cel and made by the companies Vertex and CRISPR Therapeutics, edits a gene involved in red blood cell shape and function. It appears to functionally cure the disease for at least one year. The FDA’s decision makes the U.S. the second country to approve a CRISPR therapy, following exa-cel’s approval for sickle cell disease in the U.K. in November.
Scientifically speaking, exa-cel is “an incredible asset to have,” says Michael DeBaun, a hematologist at Vanderbilt University. But it is still early to say whether the treatment will be permanent and without side effects, he adds.
On supporting science journalism
If you're enjoying this article, consider supporting our award-winning journalism by subscribing. By purchasing a subscription you are helping to ensure the future of impactful stories about the discoveries and ideas shaping our world today.
The FDA also approved another type of gene therapy for sickle cell disease today called lovo-cel, which is made by the company bluebird bio.
What is sickle cell disease, and how does the new CRISPR therapy work?
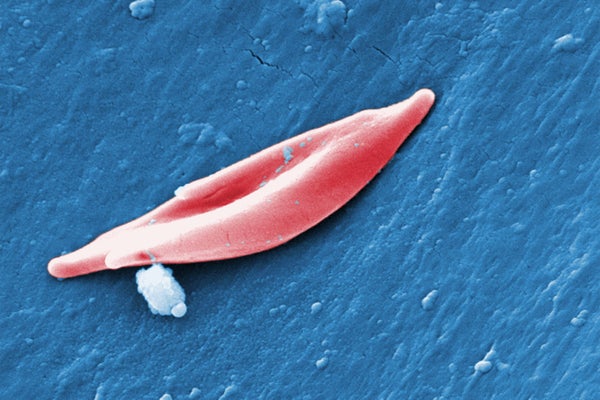
The CRISPR system used in exa-cel targets genes that produce hemoglobin, the protein that carries oxygen in blood cells. In a form of sickle cell disease called sickle cell anemia, mutations in a gene called HBB affect the protein’s structure, causing it to twist normally round red blood cells into a curved sickle shape. These sickled cells clog blood vessels, leading to severe pain and fatigue. In a related condition called beta-thalassemia, which sometimes occurs with sickle cell anemia, the body does not produce enough hemoglobin or red blood cells, resulting in symptoms caused by low blood oxygen levels. These symptoms include fatigue and growth problems in children.
Exa-cel directs an enzyme called Cas9 to a gene, called BCL11A, that typically prevents the body from making a form of hemoglobin found only in fetuses. Cas9 deactivates BCL11A in bone marrow stem cells, where red blood cells are made, by cutting its DNA, and the cells begin producing the fetal hemoglobin and creating red blood cells with a normal round shape. In the new therapy, physicians remove a person’s own bone marrow stem cells, edit them with exa-cel, destroy the rest of the person’s untreated bone marrow and then reinfuse the edited cells.
Because these edited cells eventually repopulate the body, exa-cel is considered a “curative” therapy that will theoretically last the rest of the recipient’s life, although Vertex and CRISPR Therapeutics have followed most of their trial participants for less than two years.
How effective is the treatment?
So far exa-cel has only been tested in around 100 people with either sickle cell anemia or beta-thalassemia. Nevertheless, in 2019 the FDA gave the companies a “fast-track” approval that allows it to test the therapy in smaller groups of people than would normally be required.
In these trials, which are still ongoing, 29 of the 30 study participants with sickle cell anemia had no pain for one year in the 18 months following their exa-cel transfusions. And 39 out of 42 beta-thalassemia patients no longer needed blood or bone marrow transplants—the standard treatment for this disease—for one year after the exa-cel intervention. Vertex and CRISPR Therapeutics are continuing to track the remaining participants who have not yet reached this time point and will follow all participants for up to 15 years.
What are the risks?
The results submitted to the FDA suggest that exa-cel has no major adverse health impacts, although it can cause side effects such as nausea and fever. But DeBaun points out that the participants have only been tracked for a short time and that problems could arise later.
Another concern raised by the FDA is that the Cas9 enzyme could remain active and cut the genome in places other than BCL11A, creating so-called off-target mutations. The companies modeled the most likely places in the genome where the enzyme could cut and found no evidence that this had happened in trial participants. “In light of any new therapy, we remain cautiously optimistic,” DeBaun says.
Besides exa-cel, what are some other promising therapies for sickle cell disease?
Bluebird bio's lovo-cel, the other gene therapy that was approved today by the FDA, uses a viral vector to deliver a functional version of an adult hemoglobin-producing gene and insert it permanently into a person’s genome. The data bluebird bio submitted to the FDA showed lovo-cel was effective in 36 people who were followed for a median of 32 months. Dozens of studies are investigating other types of gene therapies for sickle cell anemia and beta-thalassemia that deliver normal versions of HBB or other genes to the body.
Researchers have found that a technique called haploidentical transplant could also cure sickle cell anemia. In this treatment, which is already widely used to treat certain cancers, a person’s bone marrow cells are replaced with those from a parent or sibling who is 50 percent genetically identical to the recipient but does not have the disease. Results that will be presented next week at the American Society for Hematology’s annual conference found that 88 percent of adults who received these transplants continued to make normal red blood cells after two years. DeBaun says this technique could be particularly useful in low- and middle-income countries because it is likely to cost much less than gene editing or gene therapy.
Will exa-cel or lovo-cell be available to everyone with sickle cell disease?
Like most gene editing therapies, exa-cel and lovo-cell are likely to be very expensive. Neither Vertex, CRISPR Therapeutics nor bluebird bio have said how much their respective therapies will cost, but estimates suggest that the price for each could be as much as $2 million per patient. It is still unclear whether insurers, especially government services such as Medicaid, will cover the therapies or in which cases they will do so. Sickle cell disease disproportionately affects people of African descent, including African Americans, who are more likely to have public insurance through Medicaid than most other groups in the U.S.
DeBaun says that deciding whether to pursue CRISPR gene editing or another approach such as haploidentical transplant will need to be a shared decision by patients, their families and their physicians. Even if gene editing proves to be more effective at permanently curing the disease, it is likely to be less widely available and may take longer than a transplant from a donor.
Nevertheless, DeBaun says that exa-cel is a good first step, and he expects that the technology will improve as more is learned about CRISPR therapies over the next decade. “This is the first mile of a marathon,” he says.